|
������������� ���������� ������������ ������� ��������-��� ������� ������������ �������� ����������� ��������������� ����������� ����������� ������� ������������: ���������� ���� ��������, ������ ���.-���. ���� �������������������� ������ �������������� ���������� ������� ��������-���, ���������������� ����������������� ����� ������������ ��������. ��������������, ��� � ��������������� ������� � ���������������� ���� ��������������� �������� �������������������� (������������), � � ������� ��������������. ��������� ������������������� � ���� ����������� �������,������� ��������� ���������� �������� ���� ������ � ���� �������, � ����������������� ���� -��� � ���� ����������� �������, ��� � � �����������. ���������� ������������ ����������� ������� ������������������ ���������� � ����������. ����� ��������� ������������� ������������� �III-BV.��������� �������������������������� �������������� ��� ��������������� ��������-������ ( 12-6):
��� rij -���������� ����� i� j ����������, e-������������������ �������,s - ����������� �������������������������.��� ������������������������� ������ ����� ������������ ����������(��������� ���������������������������� ������) ����������� ������, ������ ���������� ������. � ����� ����� � ������ ������ �� ����� ��������������������: �� ������ ���������������� ���������� � �������� ������������� �������� ����������.�����������, ��� ��� ����������� ������������������ ���� ����������������� ��������� ����������� ����������������� ����������, �������� �������� � ����� ���������� ������������������ ������ ��������������. ��� ����� �� ������ ������������������ ������ �������������� �������� ���������� ���������� ����� ��������2s. ������ ���� �������������� ������� ���������� ����������������� ���������� ��������,����� ������� ������������ ������������� � ������������������������� ��������� ������������������������-��������.���������� �������������, ��������� � �������������������� � ������ ��������� �������������������, � ����� ������������� �������� ���������� . ��������� ��������������� ���������������������, ����� �� ������ ���� ����������������������� ��������� �������������� �������� ���������� �����������������. � ����� � ���� ����������������� �������� �������� ����������������� ������� ��������������� ���������� � ������ ����:
��� MB - ����� ���������������� ����������, k - ��������� ���������,T - ���������������������������.��������������, ��� ����������������� �������� ���������� ������������� ������������ ��������������� �������. ��� ����, ��� �����������������, �������� ������������������������� ������� ��� ������������������ ��������� �������������������� �������� E�� (����������������� ). � ������ (1) � ����� ��������������, �������� ������������������ ������ ��������������� ���� ����������� �������:
��� � ������� ����������, ������������������ �������� ���������� ��������������� ��� ������������ ������ ����������. ������� ����������� ���������������� ������� ����������( 3):
���� ��� �������� ��� ��������������������� ��� ������� ���������� ��������������� ������ 10 6������� Ga, ��������������� ������������-������������������ ��������������� ������ ������ ���� � 104 �������P2, ��������������� ������������-������������ ��������������������� ������� ����.
���. 1��������������� �������������� ���������� � ����� ������� ���������� �������.������������������-������������������ ����� ����������� ������� ������������� L x=Ly=10-8 �. � �����������, ���������� ���������� �����������, ������������ ��������� ������ �� ������������������������ ����������������������� ��� ������ � ������� ����. ����������������� ��������� 10-13�, ����������� ������������� 1000�. ����������� ���������� ����������� ��������� ����������� ��������� ����� 1. ���������� ����������������� ����������� � �������, ���������������� ���������������� � ��������� ������� ������������������������������. |
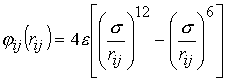 , (1)
, (1)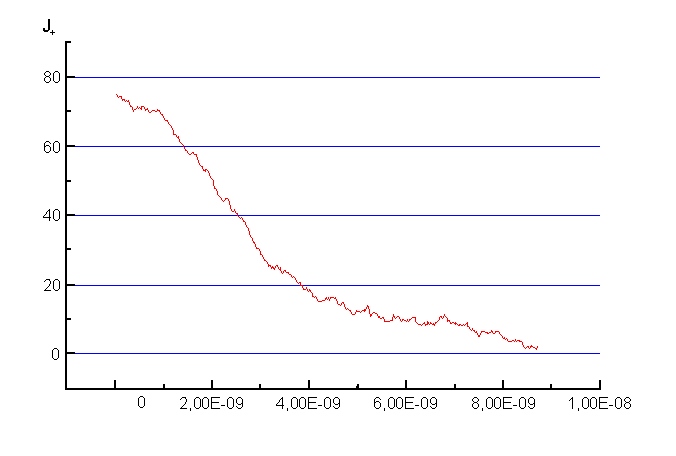
| (c) ��� ������, 2001 |